Part 1 - Getting Started
The CBM Jmol Training Guide
Introduction

But what does it really mean to "design" a protein model? It means you explore a protein structure and then simplify the way the protein is visually displayed to make the key features of the protein that help communicate your molecular story more obvious.
This can mean hiding some atoms that are not important for your protein story, changing the display format of certain parts of your protein structure or changing colors to best highlight the most important parts of the protein. Watch the video below for a more thorough description from Dr. Tim Herman.
Tim Herman, PhD Discusses What it Means to "Design" a Protein Model
Accessing Jmol
The first step in designing a molecular model is accessing Jmol
The Jmol User Design Environment (JUDE) will give you access to Jmol, and let practice all of the commands, techniques and tools described throughout this Training Guide. A link to the JUDE webpage can also be found at the bottom left of your screen throughout Jmol Training Guide.
Click to Open theJmol User Design Environment
Note: Jmol was originally a Java-based program that ran locally on your computer, rather than through a web page. For those who prefer to continue using the Java-based version of Jmol rather than the JUDE web page, we have included expandable sections with instructions specifically for Java-based Jmol users, as shown below.
Java-Based Jmol: InstallingThe Jmol User Design Environment webpage is comprised of several main areas, described in more detail below.
- The Title Bar
- The Jmol Display Window - Where your molecular structure will be displayed
- The Jmol Console - Where Jmol commands will be entered
- The Tool Column - Menus for loading molecular structures, saving designs, and reloading past work

Java-Based Jmol: Layout
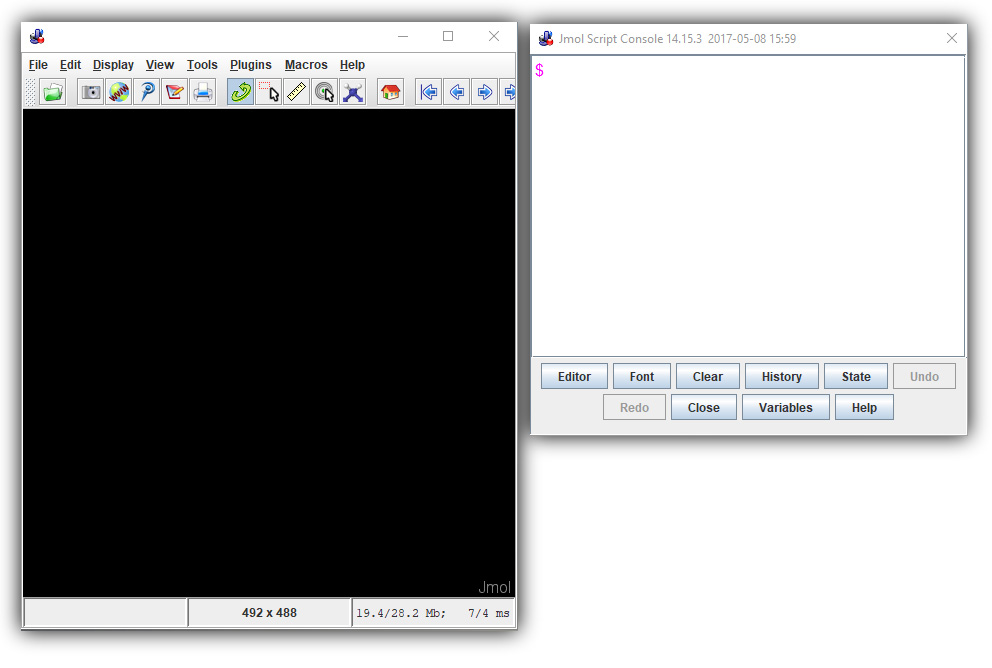
When first launched, the Java-based version of Jmol will appear simply as a black empty window. This is where the molecular rendering will be displayed once a molecular structure is loaded. But you will also need to open the Console, which is where you will enter commands that alter the appearance of your molecule.
To open the console, right click anywhere in the black Jmol window and select "Console" from the menu that appears. You will want to arrange both windows so that they do not overlap, as they will be used at the same time. Your display should look similar to the image below.
Loading Structures
In order to view a molecule in Jmol, you need a molecular structure file that contains the location in 3-dimensional space of each atom in the molecule. Every atom is assigned an Atom Number, along with an (X, Y, Z) Coordinate to mark its location.
The image below is an example of the inner contents of a molecular structure file. For more information on structure files and how they are determined, visit these RCSB Protein Databank resources:

There are two main types of molecular structure formats you can use with Jmol: Protein Databank (.PDB) files and SMILES Sequences.
SMILES Sequences - for Small Molecule Structures
SMILES (Simplified Molecular Input Line Entry Specification) Sequences are single lines of text that describe both the atoms and bonds in a small molecular structure (usually under ~50 atoms).
Atoms are represented by their element abbreviations (C, N, O, P, Cl, Br, etc.), an equals sign "=" represents double bonds and the pound sign "#" represents triple bonds. Branching is indicated by brackets "()" and rings are indicated by pairs of digits. A few examples are shown below.
- Aspirin - O=C(Oc1ccccc1C(=O)O)C
- Glucose - OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O
- Dopamine - c1cc(c(cc1CCN)O)O
SMILES Sequences can be found from a variety of online drug and small molecule databases, including the following websites.
- Wikipedia includes a SMILES sequence along the right hand column for almost all small molecule entries.
https://www.wikipedia.org/ - Drug Bank has a huge variety of resources for drugs of all kinds, including SMILES sequences for each entry.
http://www.drugbank.ca/ - ChemSpider is a free chemical structure database providing fast text and structure search access to over 34 million structures from hundreds of data sources
http://www.chemspider.com/ - Google Searches can also be a quick way to find SMILES Sequences from sources across the Internet. Simply do a search for the name of the molecule you are interested, followed by the term "SMILES".
http://www.google.com/
To open a SMILES Sequence in the CBM's Jmol User Design Environment, click the red "Open a New Molecular Structure" button to expand the load menu. Then simply type or paste the SMILES Sequence you wish to load into the first text input area and hit "Load".

Java-Based Jmol: Opening SMILES Sequences
PDB Files - for Protein Structures
Protein Databank (.pdb) files are the most common molecular structure file format for macromolecules such as proteins.
The RCSB Protein Databank (https://www.rcsb.org/) is an international database that contains over 150,000 protein structures. Each structure hosted on the Protein Databank has a unique four character long alpha-numeric identifier, referred to as the structure's PDB ID.
Protein structures on the RCSB Protein Databank can be searched by molecule name, PDB ID, or researcher name. Often more than one .pdb file will exist for a specific type of protein. For example, you will see hundreds of .pdb file entries if you search for a relatively common protein like Insulin.
The Protein Databank's Molecule of the Month (http://pdb101.rcsb.org/motm) can help identify the best .pdb files for common proteins. Each Molecule of the Month article provides an introduction to the structure and function of a specific protein, a discussion of its relevance to health and disease, and links to related entries.
Each structure on the RCSB Protein Databank has a unique PDB ID and Structure Summary page, with a variety of useful information about the structure.
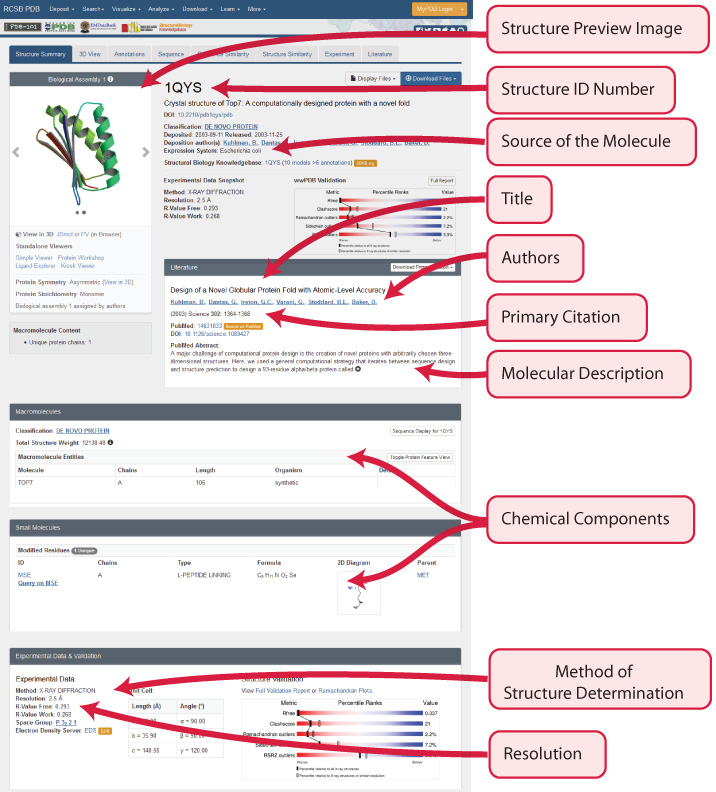
Just above the protein structure's PDB ID should be a series of tabs, the fourth of which is the Sequence Tab. This shows the exact amino acid sequence for the protein, as well as secondary structure information.

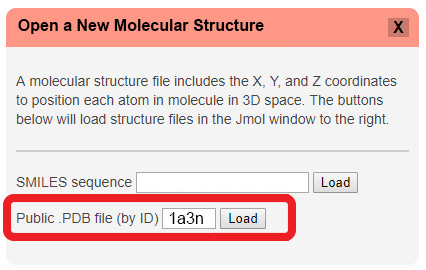
To open a PDB File in the CBM's Jmol User Design Environment, first click the red "Open a New Molecular Structure" button to expand the load menu. Then simply type or paste the four character alpha-numeric PDB-ID for the protein structure you wish to load into the second text input area and hit "Load".
Note: If you want to work with a local PDB file directly from your computer, that is not publicly available on the RCSB Protein Databank, you will need to use the Java-based version of Jmol.
Java-Based Jmol: Opening PDB FilesSaving and Reloading Your Work

Any time you design something cool in Jmol, you should save it! Saving your work allows you to revisit the design later, share the design with collaborators, and revert back to your saved state if something goes wrong.
The simplest way to save your design progress is to create a Script (.spt) file. This single file will contain all of the information needed to reproduce the current molecule representation, allowing you to open it back up exactly as it was when you saved it.
To save a .spt file in the CBM's Jmol User Design Environment, click the orange "Save Your Design" button to expand the save menu and then click the "Save .SPT File" button.
Note: Your filename must include the extension ".spt" to save properly. Also note that depending on your browser, you may need to press the "OK" button twice to initiate the save/download.

You can also save several other file formats from the JUDE Web Page, each with a specific application:
-
Image (.jpg) Files are static 2D images of your molecule design, perfect for use in PowerPoint, Word, posters or embedding in an email. To save a .jpg file, click the orange "Save your Design" button to expand the save menu, and then click the "Save .JPG File" button.
-
3D Printing (.stl) Files are the standard input filetype for most low cost plastic 3D printers, and will allow you to 3D print your own molecule designs! To save a .stl file, click the orange "Save your Design" button to expand the save menu, and then click the "Save .STL File" button.
Note: .STL files exported for 3D printing can be quite large. If your browser freezes or you receive an error when attempting to export an STL file from the JUDE Web Page, click on the section below for a work-around.
Java-Based Jmol: Saving Large STL Files(Click To Expand)XStart by saving your design as a Script (.spt) File in the Jmol User Design Environment. Click the orange "Save Your Design" button to expand the save menu and then click the "Save .SPT File" button.
Next, you will need the Java-based version of Jmol. It can be downloaded as a .zip file directly from the CBM, or you can view the full program details and version history at SourceForge.com.
Note that you will also need Java installed on your computer in order to run the Java-based version of Jmol. Click here to download Java for free.
Once downloaded, you will need to open the .zip file and copy the two files inside ("jmol.jar" and "jmol.bat") to your local hard drive. Double clicking on the .bat file for Windows or the .jar file for Mac will launch Jmol, initially showing an empty black screen.
Note: Some Mac users may receive an error, saying Jmol.jar is from an "Unidentified Developer". To allow your Mac to run Jmol.jar, you will need to open System Preferences > Security & Privacy, and then click the "Open Anyway" button near the bottom of the window.
Next, drag and drop your saved Script (.spt) File into the Jmol Display Window, which should then open your molecule design as it appeared in the JUDE Web Page.
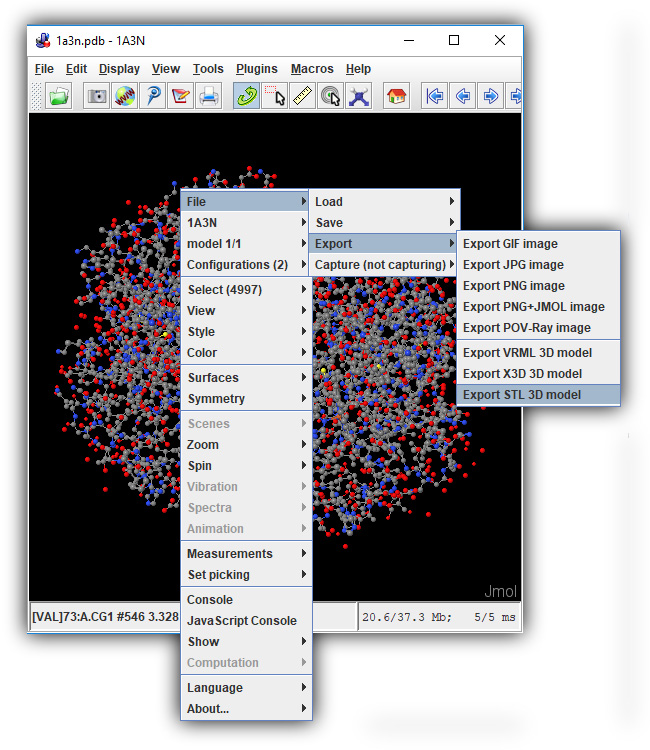
Last, right click anywhere in the Jmol Display Window and select File > Export > Export STL 3D Model from the menu that appears. The Java-based version of Jmol has access to far more memory than the JUDE Web Page, and so should be able to successfully create STL files of even very large or complicated protein designs.
For a complete guide to 3D Printing molecular models using Jmol and 3D printers, visit the 3D Printing Your Design section of the CBM's Jmol Training Guide.
-
Command History (.txt) Files are a simple text list of the commands you have entered into the Jmol User Design Environment. These can be useful in tracking your design progress, or for use in the CBM's Jmol Exploration Web Page Creator. To save a History .txt file, click the orange "Save your Design" button to expand the save menu, and then click the "Save History .TXT File" button.
Reloading a Saved .SPT File
To reload a previously saved .spt file, simply drag the file from your local computer into the Jmol Display Window in the JUDE Web Page. This will bring up your design in the Jmol Display window and allow you to continue editing.
This same drag-and-drop technique will also work if you are using the Java-based version of Jmol.
Conclusion
At this point, you should be able to access Jmol through the Jmol User Design Environment (JUDE), load SMILES and Protein Data Bank (.pdb) Files, and save and reload your work.
But that is just the beginning. . . The real power of Jmol lies in manipulating the display of your molecule. In the next section, Simplifying Your Protein, you will learn how to explore a molecular structure and view the backbone of a protein.